Phylogenetic Analysis
To analyze the evolution of Alphapapillomavirus 9 and the relationship among the HPV subtypes represented by this species, a phylogenetic tree was constructed. From 1230 genome sequence isolates obtained from the NCBI database, 90 clusters were formed using CD-Hit Suite based on an identity percentage greater or equal to 99%. Multiple Sequence alignment was done on each representative genome of 90 clusters using MAFFT online too. The result obtained was processed through MEGA 1 1 software to obtain phylogenetic trees. Neighbor-joining tree type was made using the p-distance substitution model. To further support reliability, the bootstrap method was used with the input of 100 pseudo-replicates.The Neighbor-joining method's ensuing phylogenetic tree shown below enables us to comprehend type-specific variations within genomes analyzed. Our data indicate that HPV 35 and HPV 38 have the most genomic diversity. HPV 58 and HPV 33 were closely connected. To summarize, two major groups including similar subtypes are: Group A (HPV 35, HPV 31, HPV 1 6, Alpha-9) and Group B (HPV 52, HPV 67, HPV 33, HPV 58).
click image to interact

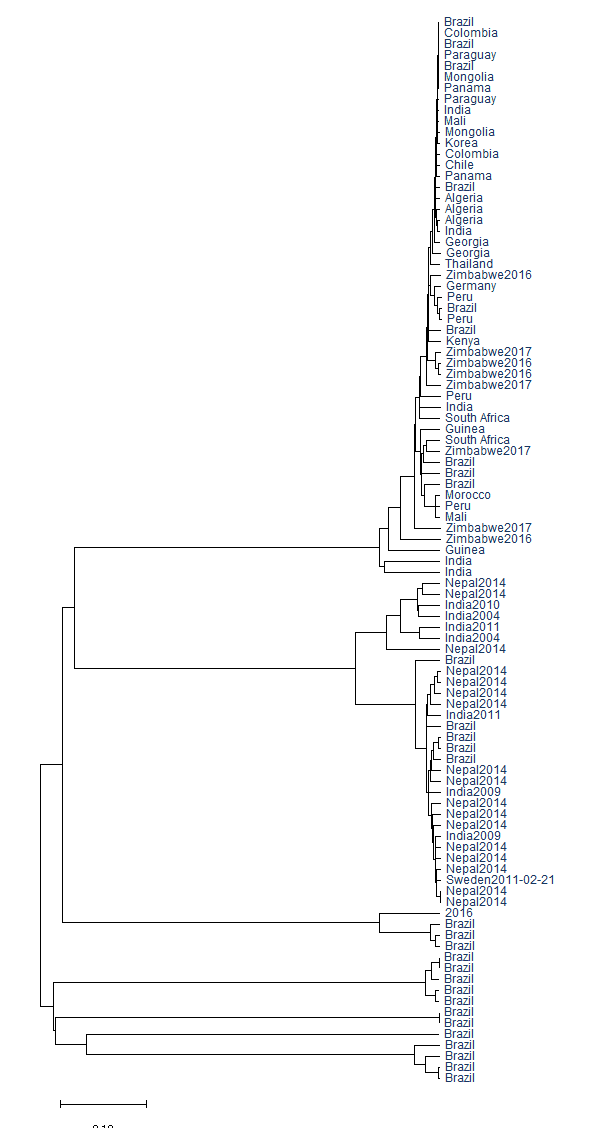
The geographical evolution of this species group was also analyzed by constructing another phylogenetic tree using the inbuilt feature at the NCBI Virus site, which matched HPV subtype evolution to their country. This tree was constructed to determine the link between isolates from various nations. 96 sequences with legitimate isolation sources were included in this study, and the resulting phylogenetic tree as shown below demonstrated that nations share comparable subtypes of species due to their geographic proximity. Additionally, the relationship between subtypes and countries was analyzed: HPV 35 is prevalent in Africa, South America, and a few Asian nations (India, Thailand, and Georgia), HPV 16 is prevalent in Brazil, India, and Nepal, while HPV 52 and HPV 33 are prevalent in Brazil.
click image to interact